TOC analysis, pure water and ultrapure water
Water purification facilities are used to generate pure and ultrapure water for pharmaceutical use via a process of pre-desalination, reverse osmosis, ion-exchange and distillation. A key parameter is the total organic carbon content (TOC). According to the guidelines of the European Pharmacopoeia, method 2.2.44, the limiting value for TOC is 500 ppb. This article describes the ‘off-line’ TOC determination of purified water with a TOC content of less than 500 ppb for robust, routine operation with practise-oriented advice.
TOC analytical results cannot be ‘better’ than those specified for the generated water product being tested. During stable continuous operation, one can expect a system-dependent quality of the purified water generated. For the purpose of this article, the quality of the water treatment facility itself is not the most important priority, rather the quality of the generated water product during regular sampling. A discussion of whether in-line or off-line TOC measurement is ‘better’ will not be discussed at this point. For this investigation, sampling forms the connecting link between the purification processing facility and the analytical system.
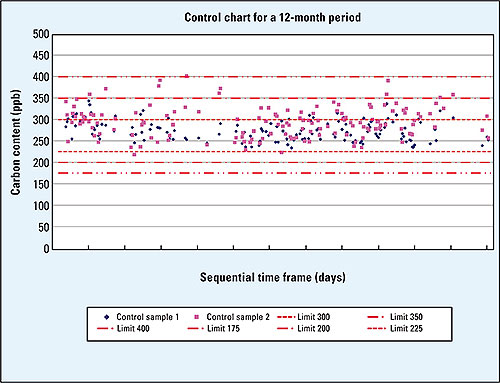 Figure 1: Example of a control chart, control samples with 250 ppb
Figure 1: Example of a control chart, control samples with 250 ppb
Robust sampling
First of all, the sampling system must be robust. The samples should be representative and must remain unchanged up to the moment of analysis. For the qualification of a water treatment facility with 10 sampling locations and with daily samplings over four weeks, this amounts to 280 samples without repetitions. The laboratory personnel should be trained accordingly. Approved sampling containers consisting of inert materials, i.e. glass with suitable seals must be available. The sampling containers must be ‘dedicated’ specifically to the use of water, pure water and ultrapure water. Intermittent use of synthetic material containers and seals for solvents such as methanol, isopropanol and ethanol excludes use of these containers for TOC trace analysis.
Critical influencing variables
Prior to sampling, an ‘initial run’ is typically specified. The valves are not disinfected prior to sampling. Furthermore, the input of air-transported carbon should be minimized, as the transfer of organic compounds from the air phase to the water phase constitutes a known contamination route. This is why the flow emanating from a valve should be laminar and the sampling containers must be ‘filled up to the mark.’ According to the available information, shelf life of samples prior to chemical analysis is not critical. Unlike microbiological assays, temporary storage in a refrigerator is not required. The dispatch chain, the total logistics after successful sampling, temporary storage, transport to the laboratory and temporary storage prior to analysis, must be tested and should be kept to a minimum.
 TOC-VCSH/N
TOC-VCSH/N
Analyses with detection limits of less than 50 ppb pose high demands on systems, processes and operating personnel. The TOC laboratory must be free from volatile organic compounds within the ppb and ppm range. Organic solvents, in particular, should not be handled in the TOC laboratory. Also, all operating personnel should refrain from using toiletries containing volatile compounds. The analysis of contaminated waters, for instance wastewaters, should not be carried out in the same room and using the same analytical instruments, so as to avoid contamination.
In the simplest case, the sample can be transferred directly from the sampling location into the sample container of the analytical system (here the ‘measuring container’, to distinguish it from the ‘sample container’ used for sampling and transport). This must be carried out under minimal laminar flow conditions using sealed measuring containers. Alternatively, the sample can be transferred from sample container to measuring container. The measuring containers must at least meet the same requirements as the sampling containers. Should the measuring containers be reused, the cleaning process must be validated. The measuring containers must be conditioned prior to use by rinsing them with ultrapure water. To summarize: the ‘ubiquitous’ contamination of samples and measuring containers and the ingress of air-transported organic compounds must be minimized.
Common TOC determination methods are UV radiolysis and catalytic combustion with subsequent IR detection of the carbon dioxide formed. The number of repeat measurements and injections up to averaging of the results can be considered as instrument-specific and programspecific, and can be assessed during each analysis. Other preconditions include qualification and calibration of the instruments to comply with the required system suitability test using benzoquinone and sucrose for the analysis of pharmaceutical water. The Pharmacopoeias describe procedures and threshold values, without making requirements for technical implementation and, in particular, for oxidation and detection:
- A system suitability test for the level of 500 ppb TOC is specified
- Limit of detection (LOD) is ≤ 0,050 ppm (≤ 50 ppb)
- ‘Reagent’ water with a TOC content of ≤ 0.100 ppm (100 ppb) is required
- ‘Reagent’ water with a conductivity of < 1.0 μS/cm at 25 °C is required
- Inorganic and organic carbon must be distinguishable from each other.
Instrument calibration
The instrument must comply with a valid calibration with known and calculated detection and determination limits according to ICH(Q)R2. The quality of TOC measurements at the trace level is also dependent on the design of the measurement sequence.
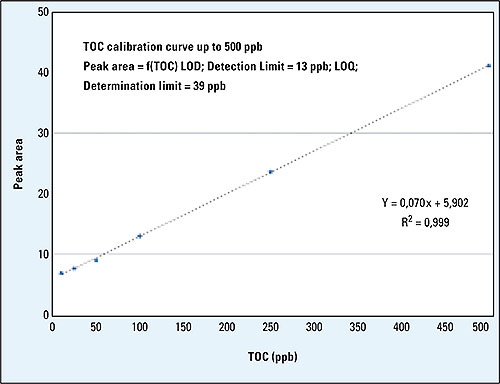 Figure 2: Example of a calibration curve, LOD and LOQ according to ICH(Q)R2
Figure 2: Example of a calibration curve, LOD and LOQ according to ICH(Q)R2
A proven method exists in which, at the start of a sequence, two samples of ultrapure water are initially measured to condition and equilibrate the flow line. While the instrument is in standby for the next measurement, substances can migrate from flow lines and valves. These are flushed from the system by the two initial measurements. A control sample containing 250 ppb TOC, followed by a control sample of ultrapure water, is subsequently measured. All other samples are then measured sequentially. This sequence must be completed with a 250 ppb control sample and an ultrapure water sample. All results are to be converted into control charts. Both 250 ppb control samples at the start and at the end of the measurement sequence, selected according to these recommendations to lie in the middle of the range of interest to the threshold value of 500 ppb, are to be considered as bracketing. In this way, it can be demonstrated that the instrument has worked properly during the entire sequence, from instrument qualification and calibration and via the implemented system suitability test up to the time of measurement. By pointing out, for instance that a catalyst change is to be carried out, this adds to the validation of TOC trace analysis.
Conclusion
The methods described enable the achievement of detection limits of 15 ppb and determination limits of 50 ppb, for instance using Shimadzu’s TOC VCPH during robust routine operation. The TOC VCPH operates under catalytic combustion conditions with subsequent NDIR detection. For the low measuring range a highly sensitive catalyst is used that allows injection volumes of up to 2 mL and enables this low detection limit. For many water analysis methods, this is a practical solution in terms of time and costs, whereby cleaning validation samples usually always more contaminated than purified water, are explicitly included. In case of cleaning validation, catalytic combustion warrants the digestion of suspended contaminated particles. It is of less importance that higher detection limits are usually specified for water in the pharmaceutical industry when compared to, for instance, the semiconductor industry. What is important here is to be able to efficiently handle strict requirements. More stringent requirements call for higher operating complexity for, in principle, a similar process.
Read for you in: GIT Labor-Fachzeitschrift 3/2010, Page 186 to 188
Dr. Wolfgang Woiwode
TECHPharm GmbH
Draisstr. 14 · 76646 Bruchsal
Germany