Analysis of residual catalysts in pharmaceuticals
ICP-OES technology quantifies 14 elements according to EMEA guidelines
 Figure 1: ICPE-9000 simultaneous inductively coupled plasma optical emission spectrometer with CCD detector
Figure 1: ICPE-9000 simultaneous inductively coupled plasma optical emission spectrometer with CCD detector
Today, the determination of toxic inorganic analytes (such as metal residues in pharmaceutical products) is a typical method for highly advanced instruments, such as inductively coupled plasma optical emission spectrometers (ICP-OES). This technology enables extremely accurate, quantifiable and reproducible analyses, and a high sample throughput.
Metal residues in pharmaceutical substances or drug products may originate from several sources, e.g. metal catalysts and metal reagents used during synthesis of the active pharmaceutical substance and the excipients, manufacturing equipment and piping, bulk packaging, the environment, cleaning solvents etc. Since metal residues do not provide any therapeutic benefit to the patient, and product risk should commensurate with the level of product benefit, the specification of a pharmaceutical substance or the drug product may need to include a limit and validated method for metal residues to guarantee acceptable product quality.
Dedicated guidelines for residues of metal catalysts
For this reason, the European Medicines Agency (EMEA), a London located decentralized agency of the European Union, has been defining dedicated guidelines, such as the guideline on specification limits for residues of metal catalysts or metal reagents in pharmaceuticals (EMEA/CHMP/SWP/4446/2000). The objective of this guideline is to recommend maximum acceptable concentration limits of metal residues arising from the use of metal catalysts or metal reagents in the synthesis of drug substances and excipients.
Since the use of these metals is restricted to defined chemical reactions, limitation of their residues in pharmaceutical substances will normally be sufficient. Limitation of these metal residues in the final drug product will normally not be necessary.
The concentration limits in this guideline are based on safety criteria and assure an adequate quality of the pharmaceutical substance and the drug product. It is therefore not considered appropriate to expect that the pharmaceutical industry tightens the concentration limits in the regulatory dossier based on GMP, process capabilities or any other quality criteria. Since the origin of metal residues is irrelevant regarding their potential toxic effects, the concentration limits in this guideline are in principle also applicable to residues from sources other than catalysts and reagents.
However, for these other sources adoption of a concentration limit and a validated method in the specification is only necessary in the very exceptional cases where these residues are known to be insufficiently limited by GMP, GDP or any other relevant provision. Pharmaceutical companies are not expected to perform extensive tests on metal residue findings of unknown sources in order to comply with this guideline. They may rely on general information from trustworthy suppliers.
The metals currently included in this guideline are listed in table 1. The guideline may be updated to include other metal residues in due course. Any interested party can submit relevant safety data. The classification and concentration limits of the currently included metals may also change when new safety data becomes available.
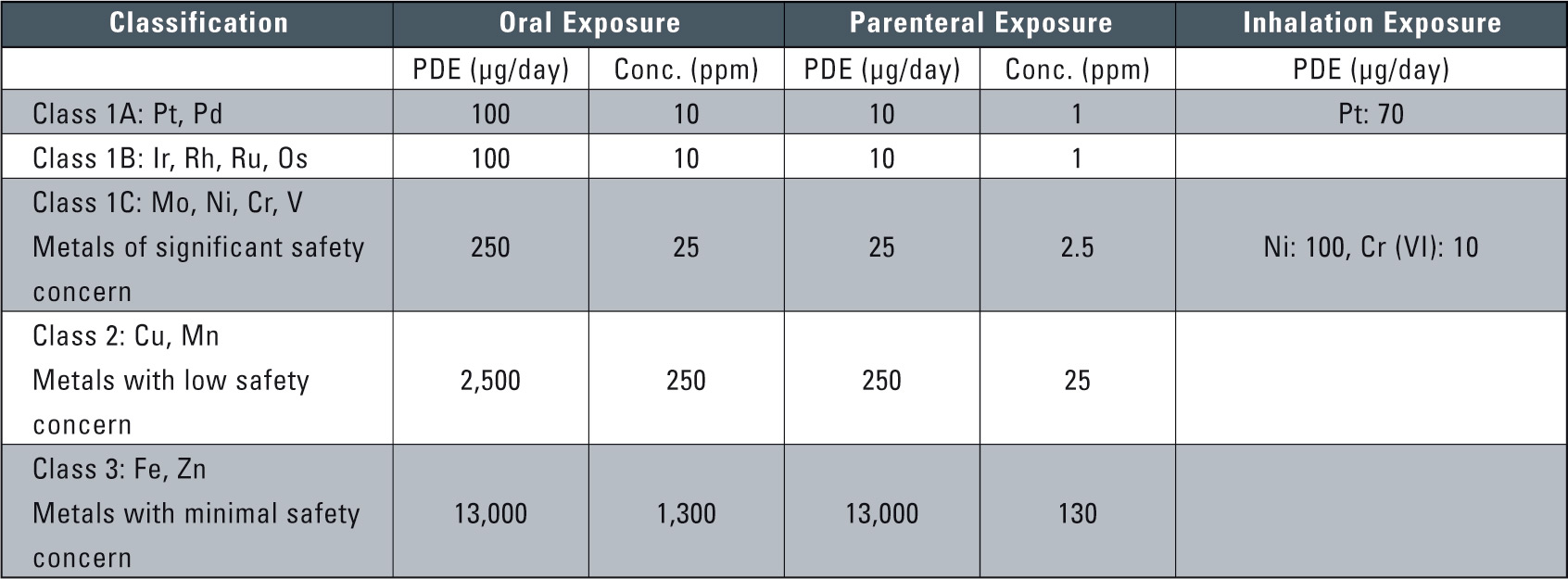 Table 1: Class Exposure and Concentration Limits for Individual Metal Catalysts and Metal Reagents (PDE: permitted daily exposure)
Table 1: Class Exposure and Concentration Limits for Individual Metal Catalysts and Metal Reagents (PDE: permitted daily exposure)
EMEA guidelines
Table 1 shows the permissible amounts and concentrations according to the guidelines issued by the EMEA on February 21, 2008. Based on risk to human health, these guidelines classified 14 types of metals into three classes. Class 1 was further divided into three sub-categories due to concerns over potential carcinogenic or other serious toxicity issues in humans. The permissible limits adressed the three administration pathways of oral, parenteral or inhalation exposure for each class. Permissible levels were based on the Guideline for Residual Solvents (ICH Q3C) 4. As the maximum permitted value for pharmaceuticals, the Permitted Daily Exposure (PDE, units µg/day) was used. The permissible concentration values were based on computing of the PDE, assuming a 10 g/day dosage of the pharmaceutical. Where daily dosage exceeds 10 g, the permissible value was computed based upon content of the active ingredients in the pharmaceutical.
Experimental work
The 14 elements of the EMEA guidelines were measured as residual metal catalysts in the pharmaceutical substances. Reagents commercially available for research purposes were used as the samples. For convenience in these tests, an organic solvent containing little contamination was selected for pretreatment of the test samples, rather than resorting to acid digestion (the choice in this case being DMSO due to its high solubility).
0.5 g of each test sample was weighed and dissolved in DMSO to bring the volume to 5 mL (a 10-fold dilution). At this time, yttrium (Y) was added as an internal standard element to produce a Y concentration of 0.1 µg/ mL, and a standard sample of tosufloxacin tosilate was added to a dilution solution to obtain a tosufloxacin tosilate concentration of 1 µg/mL. This was used for addition and for recovery testing. The solutions used to make the calibration curves were standard single-element solutions diluted with DMSO solvent. In preparing these calibration solutions, Y was also added as an internal standard element to produce a Y concentration of 0.1 µg/ mL.
1 µg/mL concentration solutions were used to prepare calibration curves and changes over time and were measured continuously over two hours. Good results were obtained; the RSD (relative standard deviation) over the two hours of measurement was less than 1 % for all elements. Furthermore, the addition and recovery samples to which the tosufloxacin tosilate was added were subjected to repeated testing over a three-day period in a reproducibility test. The results are shown in table 2.
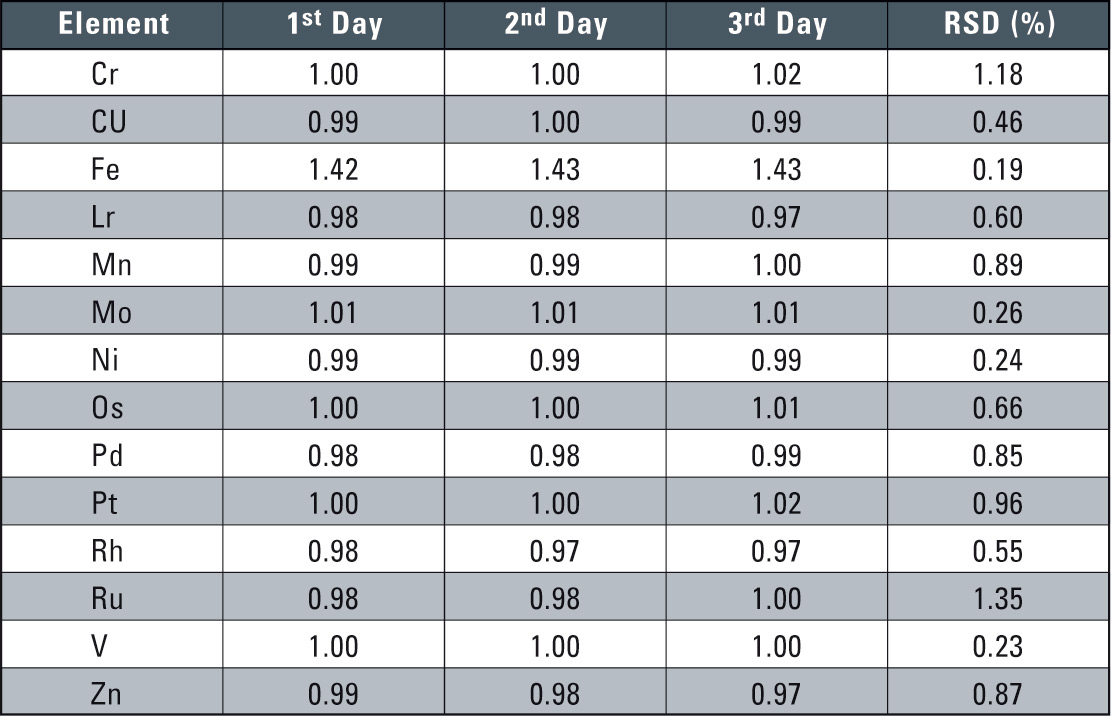 Table 2: Reproducibility Results over 3 Days (units µg/mL)
Table 2: Reproducibility Results over 3 Days (units µg/mL)
In these reproducibility tests, a calibration curve was prepared each time in performing the quantitative analysis. Very good reproducibility was obtained with each element, with the 3-day RSD being about 1 %.
Summary
In this paper, an overview of the ICP optical emission spectrometry was introduced and examples were presented using the ICPE-9000 to quantify 14 elements in pharmaceutical substances identified under EMEA guidelines, as a method for the analysis of residual catalysts in pharmaceuticals. The Japanese Pharmacopoeia (15th Revision) currently lists only the colorimetric method, a test method for heavy metals, and atomic absorption spectrometry as analytical methods for metals in pharmaceuticals. No other metal analysis methods have been adopted.
However, when reviewing the testing for metals in pharmaceuticals that has been conducted on a global scale in recent years, it is certain that the ICP analytical method will be essential for the analysis of pharmaceuticals. As demonstrated in this introduction, the ICP optical emission spectrometry is an ideal method for analyzing residual catalysts in pharmaceuticals.
References:
Director of the Evaluation and Licensing Division, the Pharmaceutical and Food Safety Bureau (Iyaku Shokuhinkyoku Shinsa Kanrika), the Ministry of Health, Labour and Welfare: Revised Guidelines on Impurities in New Drug Substances, No. 1216001, issued by the Evaluation and Licensing Division, the Pharmaceutical and Food Safety Bureau (Iyaku Shokuhinkyoku Shinsa Kanrika) December 16, 2002.
Committee for Medical Products for Human Use (CHMP): Guideline on the Specification Limits for Residues of Metal Catalysts or Metal Reagent (Doc. Ref. EMEA/CHMP/SWP/ 4446/2000). London (February 21, 2008)
USP Ad Hoc Advisory Panel on Inorganic Impurities and Heavy Metals and USP Staff: “General Chapter on Inorganic Impurities: Heavy Metals,” stimuli to the revision process, Pharmacopeia Forum 34 (5), 1345-1348 (Sept.-Oct. 2008)
International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use: ICH Harmonised Tripartite Guideline, Impurities: Guideline for Residual Solvents Q3C(R3). Step 4 version