Enthüllung der verborgenen strukturellen Vielfalt von Lipiden
Lipidisomeranalyse der nächsten Generation
Dr. Hidenori Takahashi, Shimadzu Corporation
Mit herkömmlichen MS/MS-Techniken ist es schwierig, die Positionen von Kohlenstoff-Kohlenstoff-Doppelbindungen (C=C) in Lipidmolekülen zu bestimmen, was die Unterscheidung von Strukturisomeren erschwert. Daher beschloss ein Forscher, einen neuen, effektiveren Ansatz zu entwickeln, der mit bestehenden LC-MS-Workflows kompatibel und sowohl für ESI- als auch für MALDI-Plattformen geeignet ist.
Lipide sind essenzielle Biomoleküle, die in jeder Zelle des Körpers vorkommen und sich auf die Membranstruktur, Signalübertragung und Energiespeicherung auswirken. Aus diesem Grund sind sie in verschiedenen Fachbereichen von Bedeutung, darunter Ernährung, Kosmetik, Pharmakologie und Medizin. So spielen beispielsweise das Verständnis und die Identifizierung von Lipiden eine wichtige Rolle bei der Verabreichung von Medikamenten und Therapien. Sie erleichtern die Diagnose und Beobachtung von Krankheiten, von hohen Cholesterinwerten bis hin zu Krebserkrankungen.
Lipid-Strukturisomere sind Moleküle mit derselben chemischen Formel, aber unterschiedlicher Anordnung der Atome, die durch Variationen in den Positionen der Kohlenstoff-Kohlenstoff-Doppelbindungen (C=C), den sn-Positionen und den cis/trans-Konfigurationen entstehen.[1] Insbesondere C=C-Positionsunterschiede, wie sie beispielsweise Omega-3- und Omega-6-Fettsäuren definieren, stehen mit verschiedenen biologischen Funktionen und Krankheiten in Zusammenhang.[2]
Methoden zur Analyse von Lipiden
Obwohl die Gaschromatographie-Massenspektrometrie (GC-MS) weithin zur Fettsäureanalyse eingesetzt wird, gehen bei der Derivatisierung zu Fettsäuremethylestern (FAMEs) häufig lipidklassenspezifische Informationen verloren. Da Fettsäuren aus einer Mischung aus komplexen Lipiden und freien Fettsäuren gespalten und methyliert werden, lässt sich nicht feststellen, ob die resultierenden Fettsäuren aus freien Fettsäuren oder aus bestimmten komplexen Lipiden stammen. Wenn beispielsweise die Ölsäure FA 18:1 (n-9) aus Phosphatidylcholin (PC 18:1) als Reaktion auf einen biologischen Auslöser ansteigt, während die Ölsäure FA 18:1 (n-9) aus Phosphatidylethanolamin (PE 18:1) abnimmt, lässt sich die durch GC-MS festgestellte Nettoveränderung der Ölsäure FA 18:1 (n-9) nicht genau messen.
Die Flüssigchromatographie-Massenspektrometrie (LC-MS) hingegen ermöglicht zwar eine intakte Lipidanalyse, jedoch fehlen bei der herkömmlichen energiearmen kollisionsinduzierten Dissoziation (CID) strukturelle Details, insbesondere im Hinblick auf C=C-Positionen. Um dieses Problem zu lösen, wurden neuartige Dissoziationstechniken entwickelt, darunter die ozoninduzierte Dissoziation (OzID) und elektronenbasierte Verfahren wie EIEIO (Elektronenstoßanregung von Ionen aus organischen Verbindungen).[3–4] Bei diesen Ansätzen bestehen jedoch Einschränkungen hinsichtlich der Selektivität sowie der Komplexität der Instrumente.
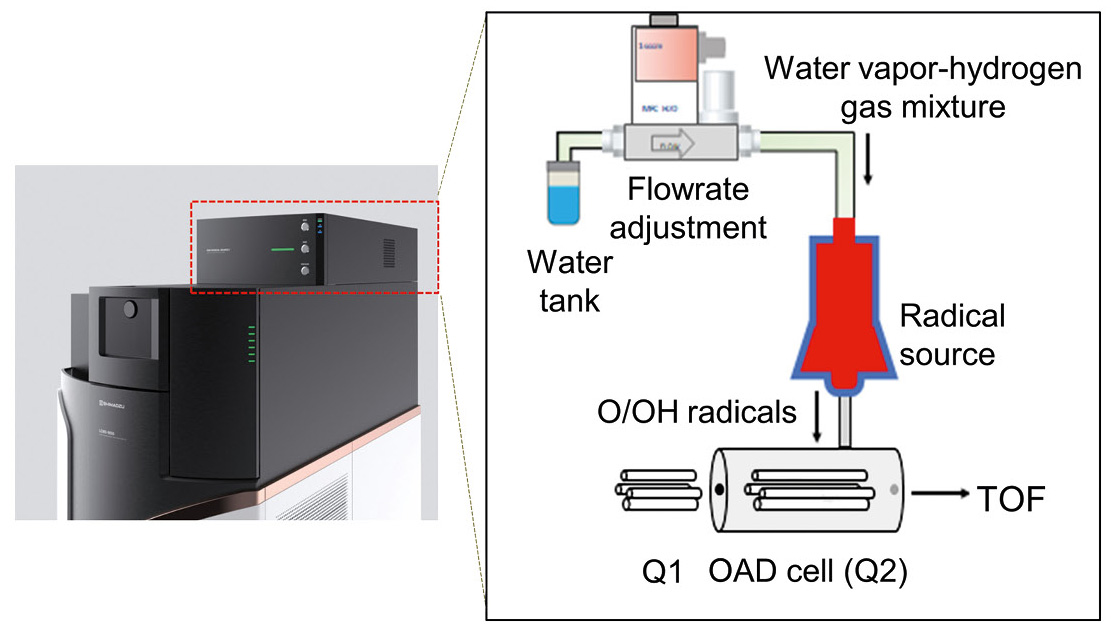
Im Shimadzu Labor von Koichi Tanaka (Nobelpreis für Chemie, 2002) wurde bereits eine neue Ionendissoziationsmethode, die Sauerstoff-Attachment-Dissoziation (OAD), entwickelt, bei der neutraler atomarer Sauerstoff (O) und Hydroxylradikale (OH•) für die radikalinduzierte Fragmentierung genutzt werden.[5] Radikale werden durch Mikrowellenentladung eines Gemisches aus Wasserdampf und Wasserstoffgas unter Vakuum erzeugt (Abbildung 1). Da O/OH•-Radikale ladungsneutrale Spezies sind, werden sie von elektrischen Feldern nicht beeinflusst und können direkt in die CID-Zelle (Q2) eines Quadrupol-Massenspektrometers eingeführt werden. Sie verändern die Ladungszustände der Ionen nicht, sodass das Verfahren weitgehend auf einfach geladene oder negativ geladene Ionen anwendbar ist. Die herkömmliche CID kann nach wie vor durch einfaches Wechseln des Kollisionsgases durchgeführt werden.
Allerdings stellte sich die Frage, ob und wie sich die OAD auf strukturelle Isomere von Lipiden anwenden lässt.
Neue Methode für ein altbekanntes Problem
Zum Nachweis der Eignung der OAD für die Lipidstrukturanalyse untersuchte ein Forscher des Tanaka-Labors menschliches Plasma (NIST SRM 1980, Millipore Sigma) nach Extraktion mittels der Bligh-und-Dyer-Methode. Die Extrakte wurden unter Stickstoff getrocknet und in Methanol rekonstituiert. Die LC-Trennung erfolgte auf einem Nexera UHPLC-System (Shimadzu) unter Verwendung einer C18-Säule (50 × 2,1 mm, 1,7 µm) bei 45 °C und 0,3 ml/min. Die mobile Phase A bestand aus ACN:MeOH:H₂O (1:1:3, v/v/v) und B aus IPA, jeweils mit 5 mM Ammoniumacetat und 10 nM EDTA. Die Gesamtanalysezeit einschließlich Säulenäquilibrierung betrug 25 Minuten; die Details entsprachen den in der Literatur beschriebenen Bedingungen.[6] Die MS-Analyse erfolgte mit einem LCMS-9050 Q-TOF (Shimadzu), ausgestattet mit der OAD Radical Source I (Shimadzu) im ESI-Modus.
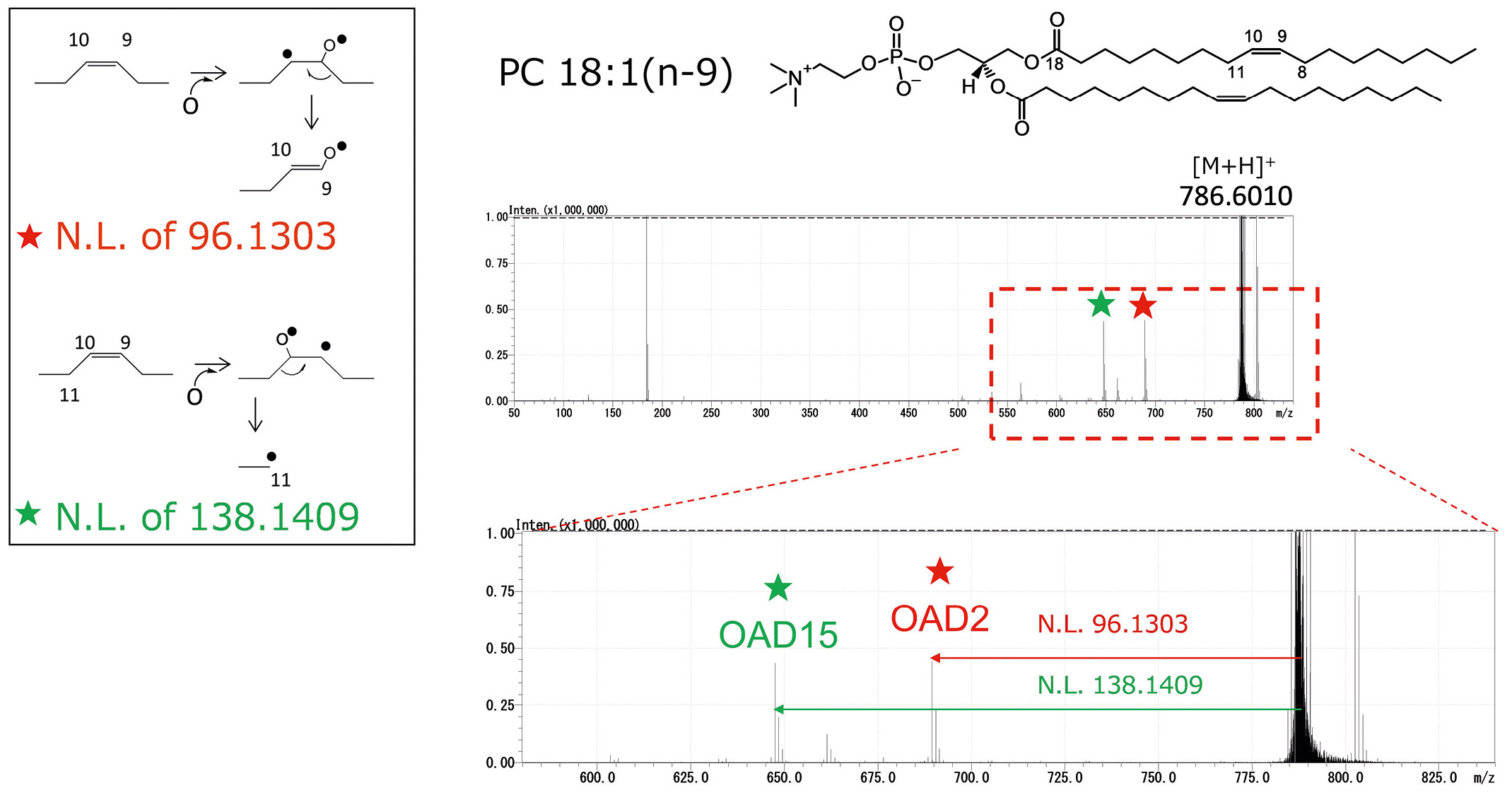
Unterscheidung von Lipidisomeren anhand der C=C-Position
In Abbildung 2 ist das OAD-MS/MS-Spektrum des Ziellipids PC 18:1(n-9)_18:1(n-9) dargestellt, wobei „PC“ die Lipidklasse Phosphatidylcholin bezeichnet, „18“ die Anzahl der Kohlenstoffatome in jeder Acylkette, „:1“ die Anzahl der C=C-Bindungen, n-9 die C=C-Position relativ zum Methylende und der Unterstrich „_“ die beiden Acylketten trennt. Die OAD induziert eine selektive Fragmentierung, die auf die C=C-Bindung abzielt, was zu zwei charakteristischen Fragmentpeaks (OAD2 und OAD15) führt, wie in Abbildung 2 dargestellt. Diese Peaks sind in herkömmlichen CID-Spektren nicht zu beobachten. Einzelheiten zur Definition und Benennung von OAD-Peaks sind in Uchino et al.[6] zu finden. In Tabelle 1 sind die neutralen Verluste zusammengefasst, die den OAD-Fragmenten für jede C=C-Position entsprechen.
Bei einer gezielten Analyse können diese vorhergesagten OAD-Neutralverluste in einer Verbindungstabelle vorab registriert werden. Extrahierte Ionenchromatogramme (XICs) können zur Unterscheidung von Strukturisomeren verwendet werden. Darüber hinaus lassen sich bei gleichzeitiger Anwendung von Kollisionsenergie (CE) bei der OAD auch neutrale Verluste vom CID-Typ bei Acylketten nachweisen.[7] Diese neutralen Verluste ermöglichen die Identifizierung der Acylkettenlänge und der Anzahl der C=C-Bindungen.[8] Somit ermöglicht die Kombination von OAD- und CID-abgeleiteten Fragmenten eine umfassende strukturelle Aufschlüsselung von Lipiden, einschließlich Informationen zur C=C-Position.
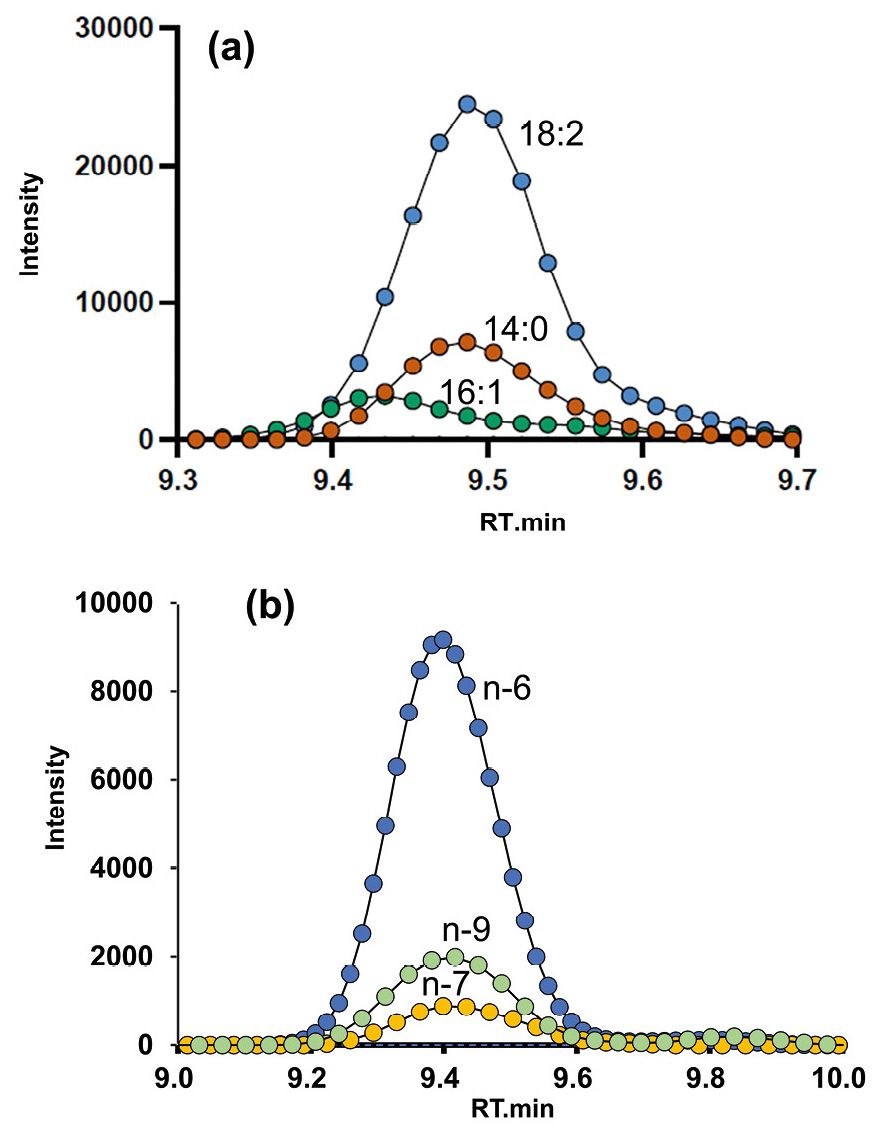
In Abbildung 3 sind die XICs von PC 32:2 ([M+H]+ = m/z 730,539) dargestellt, die unter Verwendung der OAD ermittelt wurden. „32:2“ bedeutet, dass die beiden Acylketten insgesamt 32 Kohlenstoffatome und zwei C=C-Bindungen enthalten. In Abbildung 3(a) sind die XICs für CID-basierte Acylkettenverluste dargestellt, während Abbildung 3(b) die XICs für OAD-neutrale Verluste entsprechend verschiedenen C=C-Positionen zeigt. Ausgehend von Abbildung 3(a) wurden mögliche Acylketten wie C18:2, C16:1 und C14:0 abgeleitet, mit strukturellen Kandidaten wie PC 18:2_14:0 und PC 16:1_16:1.
Anhand von Abbildung 3(b) wurden C=C-Positionen bei n-6, n-7 und n-9′ (wobei ′ das zweite C=C vom Methylende aus bezeichnet) festgestellt. Durch Kombination der Informationen aus beiden Abbildungen wurden die Acylkettenzusammensetzungen als PC 18:2(n-6, 9)_14:0 und PC 16:1(n-7)_16:1(n-7) bestimmt.
In einer früheren Studie wurden die Empfindlichkeit und die quantitative Leistungsfähigkeit der OAD unter Verwendung eines mit Deuterium markierten internen Standards, PC 15:0/18:1(d7), bewertet. Dabei wurde über einen Konzentrationsbereich von 50 fmol bis 10 pmol eine gute Linearität (R² = 0,9921) festgestellt.[7] Diese Ergebnisse zeigen, dass die OAD nicht nur hinsichtlich der Strukturanalyse hochspezifisch ist, sondern sich auch für quantitative Anwendungen eignet.
Letztendlich demonstrierte die Studie die erfolgreiche Anwendung der neu entwickelten Ionen-dissoziationsmethode OAD für die Strukturanalyse intakter Lipide. Die OAD ermöglicht die selektive und empfindliche Identifizierung von C=C-Positionen innerhalb von Lipid-Acylketten, was mit herkömmlicher CID bisher schwierig war. In Kombination mit der CID ermöglicht die OAD darüber hinaus eine umfassende Aufschlüsselung der Acylkettenzusammensetzung. Als MS/MS-basierte Methode erfordert die OAD keine Derivatisierung und lässt sich nahtlos in bestehende Analyseabläufe integrieren.
Überwindung von Herausforderungen
Erfolg kommt natürlich nicht von allein. Eine große Herausforderung bestand in der Entwicklung einer leistungsstarken Quelle für neutrale Radikale, die sich für die MS/MS eignet. Massenspektrometer werden im Hochvakuum betrieben, daher war es besonders schwierig, eine Radikalquelle zu entwickeln, die in bestehende kommerzielle Geräte integriert werden konnte, ohne die Vakuumbedingungen zu beeinträchtigen. Um dieses Problem zu lösen, wurde für diese Studie ein kompakter Radikalgenerator auf Basis von Mikrowellenentladung neu konzipiert. Eine weitere Herausforderung stellte die anfängliche Ungewissheit dar, ob die OAD-Reaktion unter Gasphasenbedingungen überhaupt stattfinden würde. Der eigentliche Durchbruch gelang daher nicht nur durch die Entwicklung des Geräts, sondern auch durch die Entdeckung einer neuen Art von chemischer Reaktion in der Gasphase.
Mehr Klarheit
Die Vorzüge der OAD zu entdecken ist, als würde man von einem Teleskop mit niedriger Auflösung zu einem mit hoher Auflösung wechseln – was zuvor wie ein einzelner Stern erschien, entpuppt sich nun als eine Ansammlung zahlreicher unterschiedlicher Sterne. Bei herkömmlichen Methoden werden Lipidisomere als ein einziges Objekt betrachtet, wohingegen die OAD ihre verborgene strukturelle Vielfalt offenbart, indem sie die Positionen der Doppelbindungen genau bestimmt. Diese verbesserte „molekulare Auflösung“ ist von entscheidender Bedeutung, da selbst geringfügige Unterschiede in den C=C-Positionen erhebliche Auswirkungen auf die biologische Funktion haben können.
| OAD2 | OAD15 | |
| n-3 | -12,0364 | -54,0470 |
| n-4 | -26,0520 | -68,0626 |
| n-5 | -40,0677 | -82,0783 |
| n-6 | -54,0833 | -96,0939 |
| n-7 | -68,0990 | -110,1096 |
| n-8 | -82,1146 | -124,1252 |
| n-9 | -96,1303 | -138,1409 |
| n-10 | -110,1459 | -152,1565 |
Diese Entwicklung ist von großem Wert für Forscher im Bereich der klinischen und pharmazeutischen Lipidomik, da die Lipidstruktur in engem Zusammenhang mit Krankheitsmechanismen, Arzneimittelwirkungen und Stoffwechselstörungen steht. Außerdem ist sie für die Lebensmittelwissenschaft relevant, wo das Verständnis der Lipidzusammensetzung und der Isomerverteilung zur Ernährungsforschung und zur Beurteilung der Lebensmittelqualität beiträgt.
Besonders hervorzuheben ist, dass die OAD auch erfolgreich in der MALDI-Bildgebungsmassenspektrometrie unter Verwendung des iMScope Systems eingesetzt wurde. Dadurch lassen sich Lipidisomere in Gewebeschnitten räumlich visualisieren, wodurch die Anwendungsmöglichkeiten der OAD über ESI-basierte Arbeitsabläufe hinaus erweitert werden. Alles in allem ermöglicht die OAD als neue Standardmethode für die Lipidstrukturanalyse viel und dürfte sowohl in der Forschung als auch in klinischen Anwendungen einen wesentlichen Beitrag zu künftigen Fortschritten in der Lipidomik leisten.
1) Harayama T., Riezman H. (2018). Membrane lipidomics for the understanding of functional membranes. Nat Rev Mol Cell Biol. 19: 281–296.
2) Han X. (2016). Lipidomics for studying metabolism. Nat Rev Endocrinol. 12: 668–679.
3) Thomas, M.C., Mitchell, T.W., Harman, D.G., Deeley, J.M., Nealon, J.R., Blanksby, S.J. (2008). Ozone-induced dissociation: Elucidation of double bond position within mass-selected lipid ions. Anal Chem. 80 (1): 303–311.
4) Baba T., Campbell J.L., Le Blanc J.C.Y., Baker P.R.S. (2016). Structural identification of triacylglycerol isomers using electron impact excitation of ions from organics (EIEIO). Anal Chem. 88 (5): 2747–2754.
5) Takahashi H., Shimabukuro Y., Asakawa D., Yamauchi S., Sekiya S., Iwamoto S. et al. (2018). Structural analysis of phospholipid using hydrogen abstraction dissociation and oxygen attachment dissociation in tandem mass spectrometry. Anal Chem. 90 (12): 7230–7238.
6) Uchino H., Tsugawa H., Takahashi H., Arita M. (2022). Computational mass spectrometry accelerates C=C position-resolved untargeted lipidomics using oxygen attachment dissociation. Commun Chem. 5 (1): 162.
7) Takeda H., Okamoto M., Takahashi H., Buyantogtokh B., Kishi N., Okano H. et al. (2025). Dual fragmentation via collision-induced and oxygen attachment dissociations using water and its radicals for C=C position-resolved lipidomics. Commun Chem. 8 (1): 148.
8) Kind, T. et al. (2013). LipidBlast in silico tandem mass spectrometry database for lipid identification. Nature methods. 10 (8): 755–758.