Bewältigung von Problemen durch Koelution mit GC-MS-Dekonvolutionssoftware
Optimierter Arbeitsablauf für mehr Identifizierungsgenauigkeit bei komplexen Gemischen
Waldemar Weber, Shimadzu Europa GmbH
Komplexe GC-MS-Proben enthalten häufig mehrere Verbindungen, die koeluieren und zu einem einzigen chromatographischen Peak verschmelzen. Diese überlagerten Spektren erschweren eine eindeutige Identifizierung, und herkömmliche Peakintegration kann die Daten leicht fehlinterpretieren. Moderne Dekonvolutionssoftware begegnet dieser Herausforderung gezielt. Dies ist besonders vorteilhaft für Labore, die mit komplexen Matrizes arbeiten, insbesondere in der Lebensmittelindustrie.
Klarer Blick auf verborgene Peaks
Labore, die mit Aromakomponenten in Lebensmitteln arbeiten oder verborgene Spurenstoffe in komplexen Proben mit hoher Matrix mittels GC-MS (Gaschromatographie-Massenspektrometrie) analysieren, benötigen häufig ein zusätzliches Werkzeug, um koeluierende Peaks zu trennen.
Das leistungsfähigste und effizienteste Werkzeug hierfür ist Dekonvolutionssoftware. Sie löst die Herausforderung, indem sie das gemessene Signal als Überlagerung einzelner Elutionsprofile modelliert. Das Ergebnis sind ein aufgelöstes Dekonvolutionschromatogramm, das mit dem Totalionenchromatogramm (TIC) abgestimmt ist, die Trennung verborgener Komponentenpeaks sowie die Rekonstruktion sauberer MS-Spektren für jede einzelne Verbindung. Insgesamt erhöht dies die Sicherheit bei der qualitativen Analyse von Verunreinigungen und Spurenstoffen, die durch stark vertretene Analyten überdeckt werden.
Durch aufgelöste Spektren für jede Komponente können Analytiker Identitäten transparenter überprüfen, indem sie objektive Spektrenabgleiche nutzen und mehrere passende Kandidaten mit klarer Ähnlichkeit in der Bibliothek der Dekonvolutionssoftware bewerten. Gleichzeitig unterstützt die Dekonvolution gezielte Arbeitsabläufe: Bekannte Verbindungen lassen sich schnell screenen, um den Suchraum einzugrenzen, während unbekannte Stoffe mithilfe von Spektrenbibliotheken untersucht werden.
Praxisbeispiel
Zur Veranschaulichung der Vorteile von Dekonvolutionssoftware in der GC-MS-Analytik wurde eine kombinierte Mischung aus Pestizidstandards (Mix 64, Mix 13 und Mix 7) mit insgesamt 50 Verbindungen untersucht. Die Datenerfassung erfolgte mit einer Niederdruck-GC-Methode, die den Strömungswiderstand reduziert und dadurch sehr kurze Laufzeiten ermöglicht, jedoch auf Kosten der chromatographischen Trennleistung. Auf diese Weise wurden gezielt Koelutionen erzeugt. Im Folgenden wird gezeigt, wie die Dekonvolution diese überlagerten Peaks zuverlässig trennt und saubere Spektren für einzelne Pestizide rekonstruiert.
Original-TIC und Dekonvolutionschromatogramme sowie repräsentative Spektren und praxisnahe Einstellungen zeigen, wie beschleunigte GC-Workflows hochpräzise qualitative Ergebnisse liefern können.
Analytische Hardware und Software:
Haupteinheit: Nexis GC-2030 mit QP2050 Massenspektrometer
Zubehör: AOC-30i Flüssigsampler
Hauptverbrauchsmaterialien: SH-5MS LPGC-Säule (5 m × 0,18 mm × 0,01 μm)
Software: LabSolutions GCMS und LabSolutions Insight Explore
Erreichen der erforderlichen Klarheit
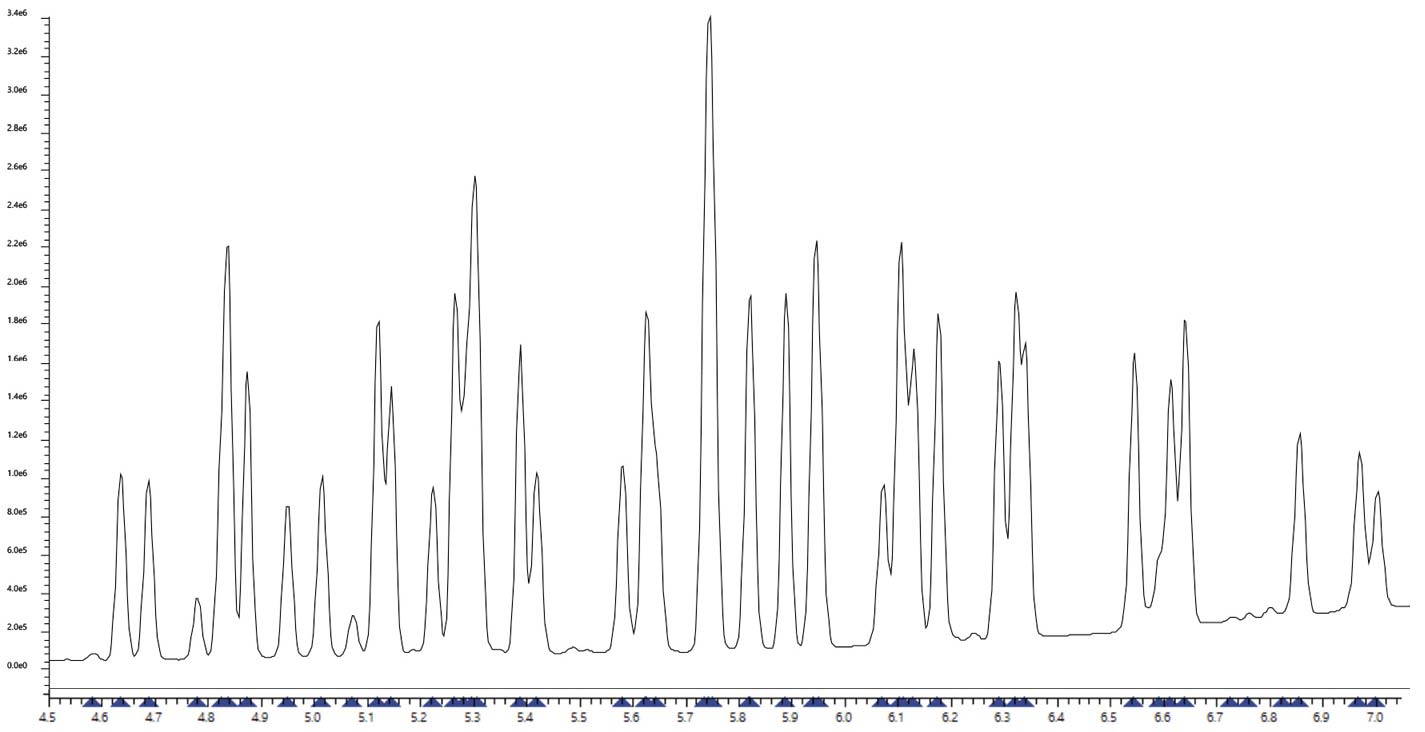
Die Ergebnisse für die untersuchte Pestizidmischung sind in Abbildung 2 dargestellt. Das Chromatogramm zeigt den Gesamtionenstrom des eingesetzten Gemischs. Blaue Dreiecke auf der x-Achse kennzeichnen die von der Dekonvolutionssoftware erkannten Koelutionen. Der Niederdruck-GC-Ansatz ermöglicht sehr schnelle Messungen, in diesem Fall etwa 7,5 Minuten. Ein häufiger Nachteil ist jedoch die eingeschränkte Trennleistung. Obwohl das Gemisch aus 50 Pestiziden weniger komplex ist als typische Screening-Mischungen, ist die Anzahl der Koelutionen hoch, was eine eindeutige Identifizierung erschwert und unsicher macht.
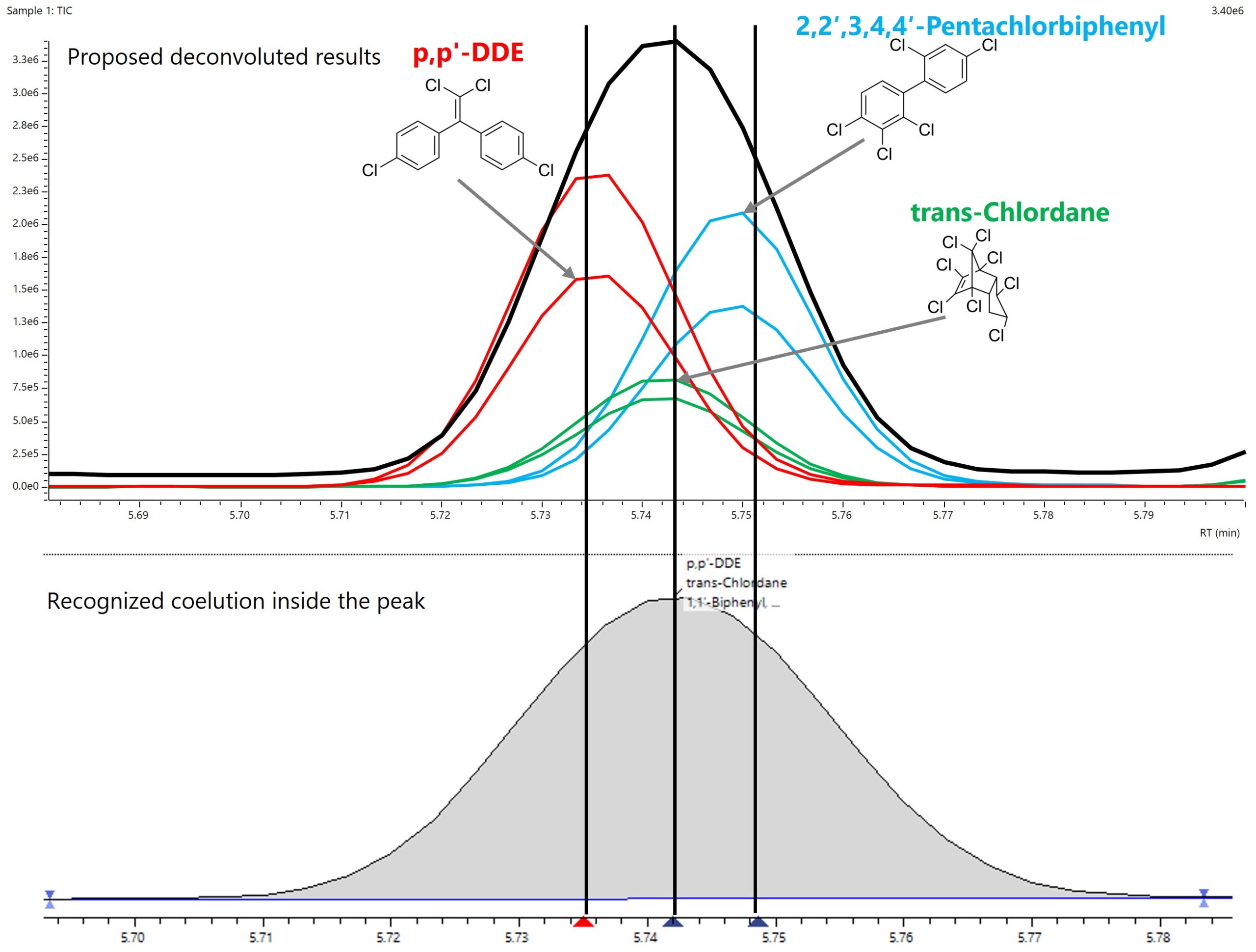
Für das qualitative Screening gezielter Verbindungen bietet die Kombination aus Niederdruck-GC-MS und anschließender Dekonvolution jedoch sowohl Geschwindigkeit als auch Genauigkeit. Ein Beispiel ist ein Peak bei einer Retentionszeit von 5,74 Minuten, dargestellt in Abbildung 3.
Abbildung 2 zeigt, dass an dieser Stelle drei Verbindungen gemeinsam eluieren: p,p’-dde, 2,2’,3,4,4’-Pentachlorbiphenyl und trans-Chlordan. Der obere Teil der Abbildung veranschaulicht die Auftrennung durch Dekonvolution. Für jede Verbindung sind zwei charakteristische m/z-Werte dargestellt: p,p’-dde mit m/z 246 und 248, Pentachlorbiphenyl mit m/z 373 und 375 sowie trans-Chlordan mit m/z 326 und 328.
Die Dekonvolution mit dem LabSolutions Insight Explore Modul trennt alle drei Komponenten effektiv und ermöglicht eine zuverlässige Identifizierung. Eine herkömmliche Hintergrundkorrektur entfernt häufig nur teilweise störende Massenfragmente aus dem Spektrum, was zu mehr falsch positiven und falsch negativen Ergebnissen führen kann. Am Beispiel des Peaks bei 5,74 Minuten zeigt Abbildung 3, dass eine einfache Hintergrundkorrektur im Spektrum von p,p’-dde fehlerhafte Fragmente bei m/z 372 bis 373 enthält, die eindeutig von trans-Chlordan stammen. Das dekonvolvierte Spektrum stimmt dagegen sehr gut mit dem Referenzspektrum überein. Eine Bibliothekssuche im Anschluss an die Dekonvolution liefert daher deutlich präzisere Ergebnisse.
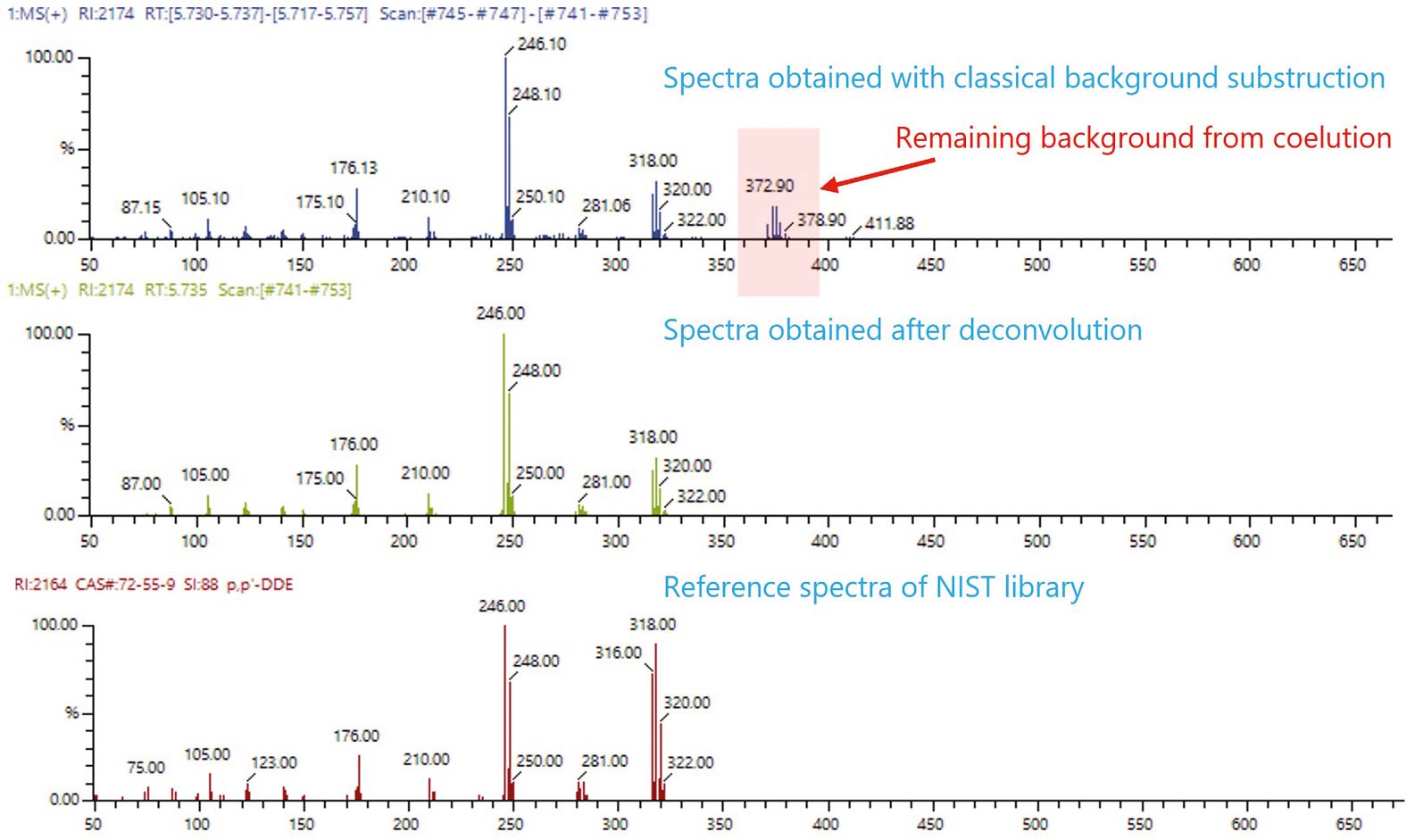
Diese Anwendung zeigt, dass Dekonvolution die qualitative GC-MS-Analyse komplexer Chromatogramme mit kurzen Laufzeiten deutlich verbessert, indem sie koeluierende Verbindungen trennt und für jede Komponente saubere Massenspektren rekonstruiert. In Kombination mit einer auf Geschwindigkeit optimierten Niederdruck-GC-Methode lassen sich mittels Dekonvolution überlagerte Pestizidsignale zuverlässig auflösen, was mit konventioneller Integration und einfacher Hintergrundkorrektur nicht möglich ist. Die Übereinstimmung mit Bibliotheksspektren wird verbessert und die Anzahl falsch positiver sowie falsch negativer Ergebnisse reduziert. Dadurch werden schnelle und verlässliche Screening-Workflows ermöglicht, bei denen Zielverbindungen effizient analysiert und unbekannte Substanzen mit höherer Sicherheit identifiziert werden können.