Offenbaren Ihre Peaks Daten – oder verbergen sie diese?
Dr. Anna Cooper, Shimadzu UK
Expertenberatung zur Fehlerbehebung: Tipps für präzisere HPLC-Peaks
Labore verwenden bei ihrer Arbeit hochsensible und hochentwickelte Instrumente. Doch diese Instrumente – wie technologisch fortschrittlich sie auch sein mögen – sind nach wie vor Geräte und verhalten sich mitunter auch so. Hinzu kommt, dass Laboranwender häufig mit der Entwicklung neuer Methoden befasst sind: Sie setzen ihre Instrumente auf ungewöhnliche und neuartige Weise ein, um immer bessere Ergebnisse zu erzielen, und testen dabei die Grenzen der Leistungsfähigkeit ihrer Geräte aus.
Laboranwender wissen sehr gut, dass die Fehlerbehebung ein fester Bestandteil der täglichen Laborarbeit ist. Häufig ziehen sie dabei die Experten von Shimadzu zurate. Mit diesem Artikel beginnt eine neue „Secrets of Science“-Reihe zum Thema Fehlerbehebung bei Analysegeräten. Konkret geht es in diesem Beitrag um die Vermeidung und Behebung von Problemen mit der Peakform in der Hochleistungsflüssigkeitschromatographie (HPLC).
Fehlerbehebung bei Problemen mit der Peakform in der HPLC
Eine gute Peakform ist für eine zuverlässige Chromatographie unerlässlich. Die ideale chromatographische Peakform ist gaußförmig mit symmetrischem Anfang und Ende des Peaks. In der Praxis wird dies jedoch aus verschiedenen Gründen, etwa wegen sekundärer Wechselwirkungen oder alternder stationärer Phasen, häufig nicht erreicht. Der Symmetriefaktor sollte zwischen 0,8 und 1,2 liegen, um die Wahrscheinlichkeit zu erhöhen, dass die relevanten Peaks aufgelöst werden und Verunreinigungen nicht unter dem Peak verborgen bleiben. Jede Abweichung von den erwarteten Peakprofilen kann auf Probleme mit dem System, der Methode oder der Säulenleistung hinweisen, die letztlich die Auflösung, Quantifizierung und Datenintegrität beeinträchtigen. Da die Peakform von vielen miteinander verbundenen Faktoren beeinflusst wird, ist bei der Fehlerbehebung ein systematischer Ansatz erforderlich, mit dem sowohl hardware- als auch methodenbezogene Ursachen berücksichtigt werden. Ziel ist es, spitzere Peaks und damit eine bessere Datenqualität zu gewährleisten.
Häufige Ursachen für Probleme mit der Peakform
Stärke des Probenlösungsmittels
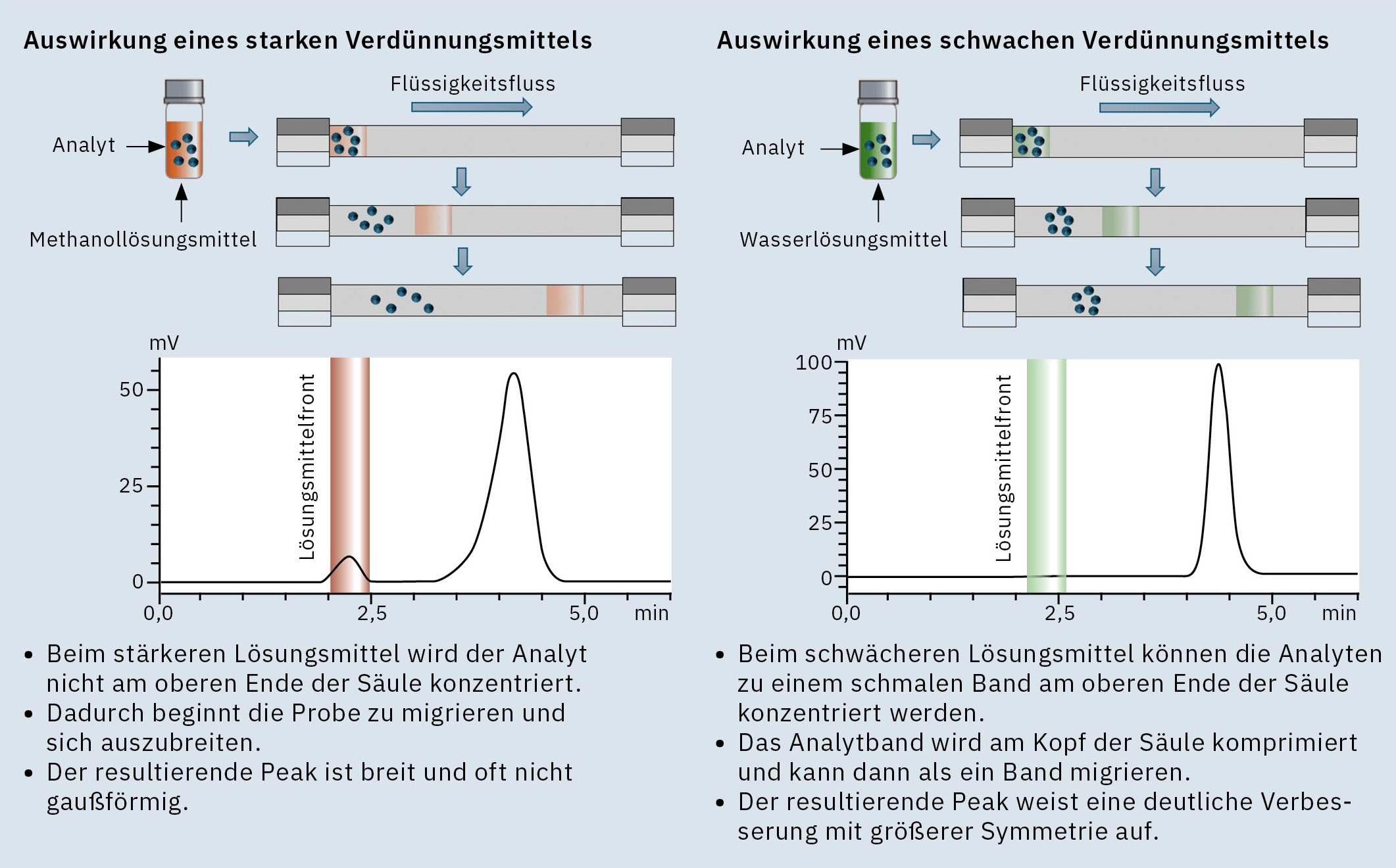
Die Zusammensetzung des Probenlösungsmittels spielt eine wesentliche Rolle für die Peakform. Idealerweise sollte sie den Ausgangsbedingungen der chromatographischen Methode möglichst genau entsprechen, um Störungen zu minimieren.
- Schwächere Lösungsmittel (z. B. Wasser in der Umkehrphasen-HPLC) können die Peakschärfe verbessern, indem sie die Analyten am Säulenkopf konzentrieren.
- Starke Lösungsmittel (z. B. 100%iges Methanol oder Acetonitril) bergen das Risiko einer Peakverbreiterung oder sogar -aufspaltung, da die Analyten vom Lösungsmittel mitgetragen werden können, anstatt effektiv zurückgehalten zu werden. Dies ist besonders problematisch bei früh eluierenden Peaks, deren Peakform durch ein starkes Verdünnungsmittel beeinträchtigt wird (Abbildung 1).
Lösungen
- Das Probenlösungsmittel sollte nach Möglichkeit mit der anfänglichen mobilen Phase abgestimmt werden.
- Wenn die Probe nicht in einem geeigneteren Lösungsmittel gelöst werden kann, kann eine Koinjektion mit Wasser („Sandwich“-Injektion) durchgeführt werden. Hier wird der Autosampler so programmiert, dass er zunächst ein bestimmtes Volumen Wasser ansaugt, dann die Probe und anschließend erneut Wasser, um eine konzentrierte Probe am Kopf der Säule zu erzeugen und so die nachteiligen Auswirkungen der Bandbreitenverbreiterung zu neutralisieren.
Zu hohes Injektionsvolumen oder zu viel Lösungsmittel
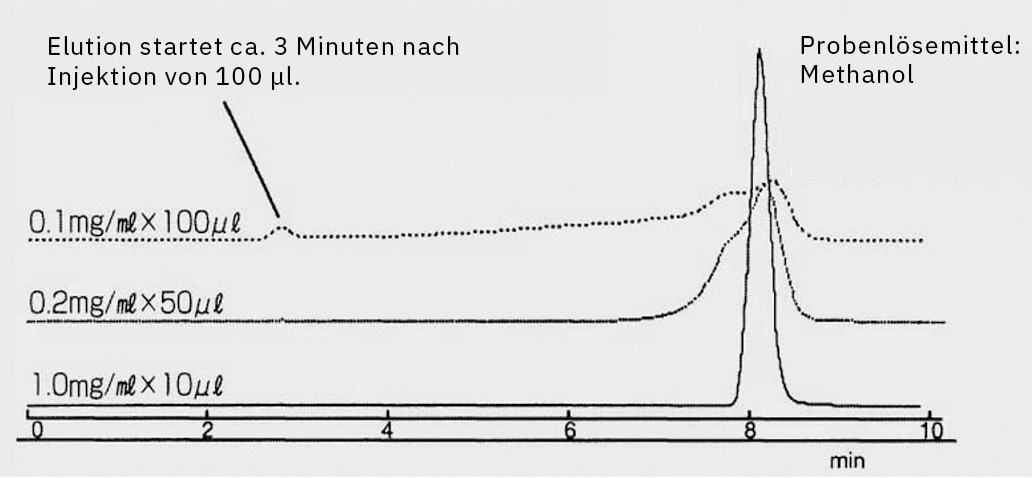
Selbst bei Verwendung eines geeigneten Lösungsmittels verzerren zu hohe Injektionsvolumina die Peaks und verringern die Effizienz. Wenn zu viel Probe injiziert wird, sind alle aktiven Stellen am Säulenkopf belegt und die verbleibende Probe fließt mit verminderter Wechselwirkung an den belegten Stellen vorbei. Durch die Überladung werden die aktiven Stellen der stationären Phase – insbesondere am Säuleneingang – gesättigt, was je nach Analyt zu sogenanntem Peak-Fronting oder -Tailing führt.
Darüber hinaus können sich durch die Überladung der Säule die Retentionszeiten verschieben und die Peaks verbreitern. Dies ist insbesondere bei der Hochskalierung problematisch: Eine Verbindung, die unter analytischen Bedingungen gut aufgelöst wird, kann sich unter überladenen präparativen Bedingungen verschieben und verbreitern, was zur Folge haben kann, dass kleinere Peaks von Verunreinigungen koeluieren und unentdeckt bleiben.
Wenn hohe Injektionsvolumina unvermeidbar sind, sollte eine Säule mit größerem Innendurchmesser oder höherer Beladungskapazität in Betracht gezogen werden, um diese Effekte zu minimieren (Abbildung 2).
Lösungen
- Verringerung des Injektionsvolumens, sofern die Empfindlichkeit dies zulässt
- Konzentration der Proben, um kleinere Injektionsvolumina zu ermöglichen, ohne die Detektion zu beeinträchtigen
- Verwendung von Säulen mit höherer Beladungskapazität oder größerem Innendurchmesser, wenn Injektionen mit hohem Volumen unvermeidbar sind

Datenaufnahmeraten und Detektoreinstellungen
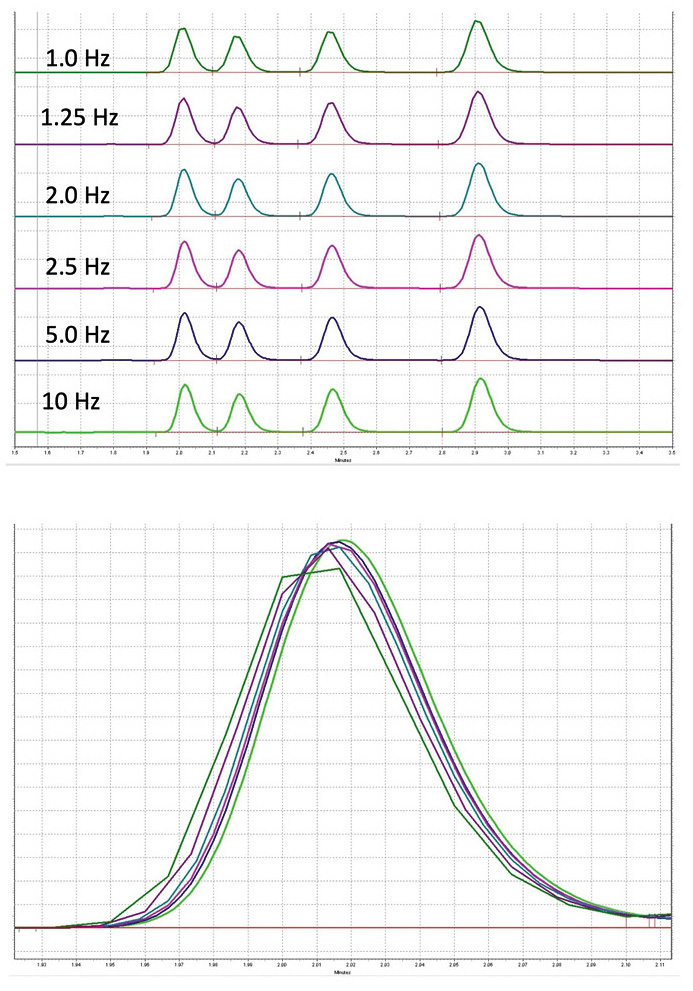
Ein häufig übersehener Faktor in der Chromatographie ist die Datenaufnahmerate, die einen erheblichen Einfluss auf die Qualität des Chromatogramms hat. Die Anpassung der Aufnahmerate an die jeweilige chromatographische Methode ist daher unerlässlich. Sie beeinflusst nicht nur das Erscheinungsbild des Chromatogramms, sondern auch die Genauigkeit der Daten.
Mit einer niedrigen Datenaufnahmerate gemessene Peaks werden aufgrund einer zu geringen Anzahl von Datenpunkten unzureichend beschrieben. Dies kann erhebliche Schwankungen in der Peakfläche hervorrufen, da geringfügige Abweichungen dazu führen können, dass kein Datenpunkt das tatsächliche Peakmaximum erfasst. Umgekehrt kann eine zu hohe Datenerfassungsrate ebenfalls Nachteile mit sich bringen, etwa ein erhöhtes Rauschen, das kleinere Peaks überdecken oder die allgemeine Signalklarheit beeinträchtigen kann, sowie zu sehr großen Datendateien führen.
Auf den ersten Blick erscheinen die Peaks im Bild rechts oben nahezu identisch, obwohl sie mit unterschiedlichen Datenaufnahmeraten gemessen wurden. Bei Überlagerung der Chromatogramme und Vergrößerung des Ausschnitts, wie in der Abbildung rechts unten dargestellt, werden die Unterschiede jedoch deutlich (Abbildung 3).
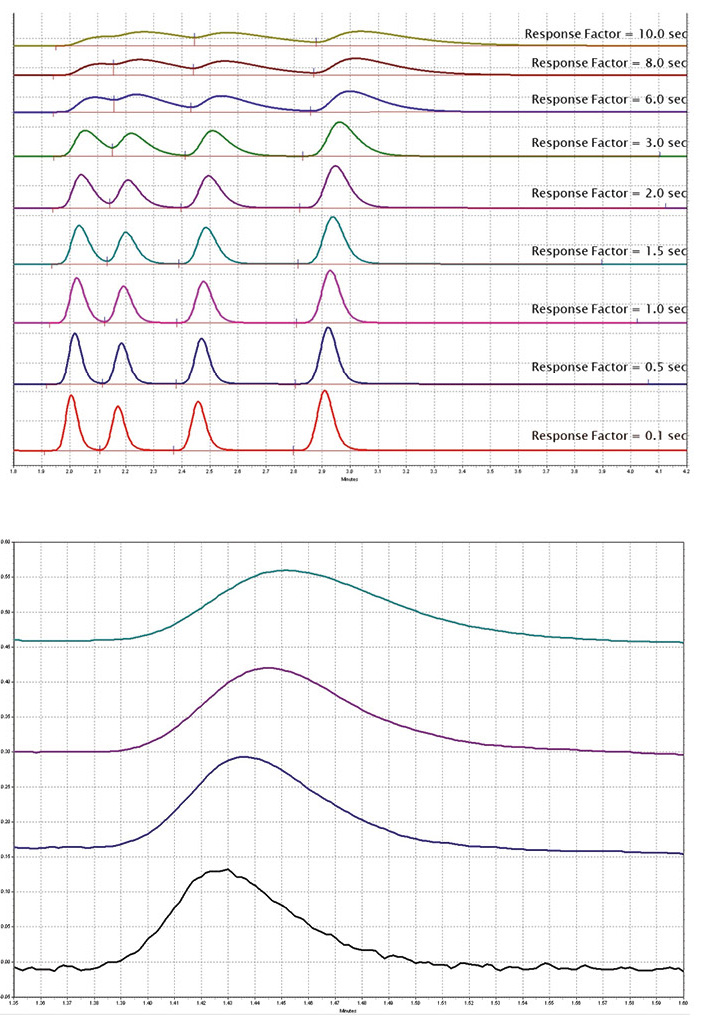
Ein weiterer Parameter ist die Empfindlichkeitseinstellung des Detektors. Ist die Empfindlichkeit zu hoch eingestellt, werden die Peaks künstlich verbreitert, was zu einer Verringerung der Auflösung führt. Bei einer zu niedrigen Empfindlichkeitseinstellung werden die Peaks hingegen verengt, jedoch das Rauschen verstärkt (Abbildung 4).
Lösungen
Die schnellste Aufnahmerate und die niedrigste Empfindlichkeitseinstellung, die noch ein annehmbares Signal-Rausch-Verhältnis ermöglichen, sollten verwendet werden. Diese Einstellungen sollten während der Methodenentwicklung optimiert werden, anstatt sich auf die Standardparameter des Geräts zu verlassen. Bei UHPLC-Methoden ist für eine genaue Beschreibung der Peaks eine höhere Aufnahmerate erforderlich; sie liegt in der Regel über 12,5 Hz.
Praktische Lösungen für spezifische Probleme mit der Peakform
Peak trailing
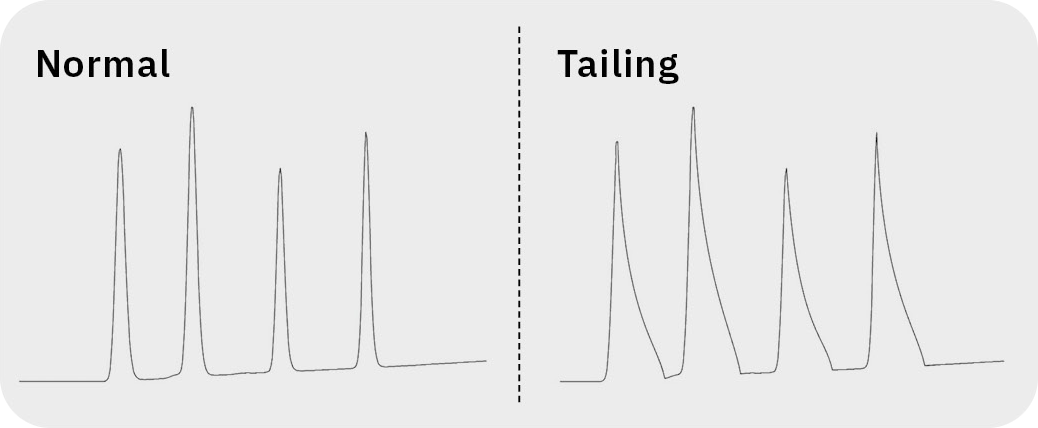
Eine der häufigsten Veränderungen der Peakform ist das sogenannte Tailing, bei dem die hintere Hälfte des Peaks breiter ist als die vordere und sich nach hinten verlängert. Die Ursache hierfür liegt häufig darin, dass der Analyt mehr als einen Retentionsmechanismus auf der Säule aufweist.
Bei der Umkehrphasenchromatographie erfolgt die Retention des Analyten jedoch hauptsächlich durch hydrophobe Wechselwirkungen, sodass es häufig zu synergistischen Mechanismen innerhalb der Säule kommt. Bei ionisierten basischen Verbindungen kann beispielsweise die positive Ladung mit den freien Silanolgruppen auf dem Siliziumdioxid wechselwirken, was zu Tailing führen kann. Da Silanolgruppen sauer sind, lassen sich Wechselwirkungen mit dem Analyten durch Senkung des pH-Werts der chromatographischen Bedingungen minimieren. Sollte dies nicht möglich sein, empfiehlt es sich, die Säule durch eine deaktivierte Säule zu ersetzen, beispielsweise eine Polymersäule, eine Säule mit guter Endkappung oder eine Säule mit leicht positiver Oberflächencharakteristik. Einige Säulenhersteller (z. B. Shimadzu) bieten spezielle Säulen für die Analyse stark basischer Substanzen an.
Eine beschädigte Säule oder falsche Analysebedingungen, etwa ein falscher pH-Wert der mobilen Phase, können ebenfalls zu Tailing führen (Abbildung 5).
Lösungen
- Durch eine Senkung des pH-Werts der mobilen Phase lässt sich die Silanolaktivität unterdrücken.
- Der Wechsel zu endgekappten oder polymerbasierten Säulen, die zur Minimierung von Silanolwechselwirkungen entwickelt wurden, ist ebenfalls eine Option.
- Der pH-Wert und die Bedingungen der mobilen Phase müssen innerhalb der Spezifikationen der Säule liegen.
- Außerdem sollte die Ionenstärke des Puffers erhöht werden, um silanophile Wechselwirkungen zu reduzieren.
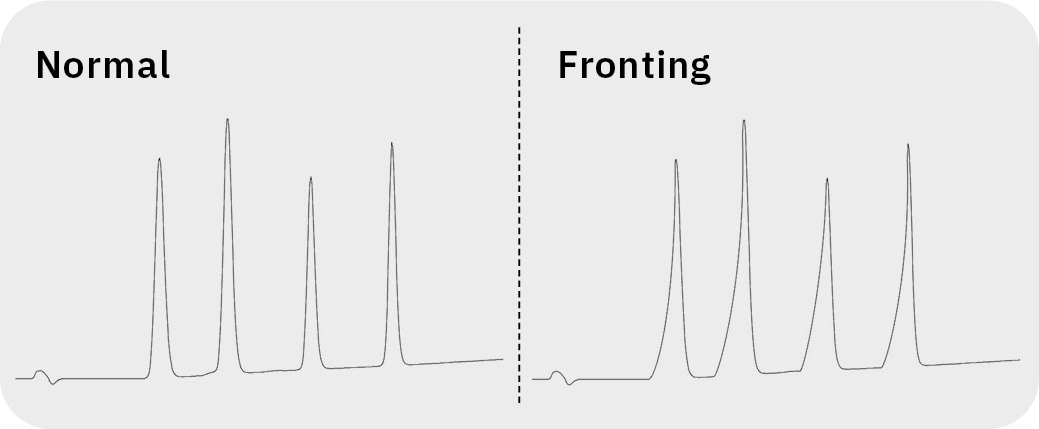
Peak fronting
Fronting beschreibt die gegenteilige Asymmetrie des Peaks beim Tailing, bei dem die Vorderkante breiter als das Ende ist und einer Haifischflosse ähnelt. Bei allmählich zunehmendem Fronting ist in den meisten Fällen eine defekte Säule die Ursache. Die stationäre Phase kann durch natürliche Alterung oder durch die Verwendung der Säule außerhalb der Spezifikationen (z. B. Temperatur oder pH-Wert) beschädigt werden. In diesem Fall helfen nur der Austausch der Säule und die Anpassung der chromatographischen Bedingungen.
Wie bereits erwähnt, kann jedoch auch eine Überladung der Säule zu Fronting führen. Die Reduzierung des Injektionsvolumens oder die Verwendung einer Säule mit höherer Kapazität bieten hier Abhilfe.
Eine weiterer Grund für Peak-Fronting kann ein inkompatibles Probenlösungsmittel oder eine schlechte Löslichkeit der Probe in der mobilen Phase sein. In diesem Fall muss lediglich das Probenlösungsmittel gewechselt werden. Eine zu niedrige Säulentemperatur kann ebenfalls zu Fronting führen (Abbildung 6).
Lösungen
- Die Probenmenge sollte reduziert werden.
- Wenn möglich, sollte eine Säule mit höherer Kapazität verwendet werden.
- Eine gute Lösungsmittelverträglichkeit sollte sichergestellt werden.
- Die Temperaturregelung sollte überprüft werden.
- Wenn das Fronting allmählich zunimmt, kann dies an einer Alterung der Säule liegen: In der Regel hilft nur ein Austausch.
Peakverbreiterung
Bei isokratischen Trennungen ist zu erwarten, dass sich die Analytbande bei der Migration durch das Säulenbett in Abhängigkeit von der Verweildauer in der Säule verbreitert. Dies führt zu breiteren Peaks bei spät eluierenden Verbindungen. Anhand dieses Faktors lässt sich sicherstellen, dass der Analyt ausreichend mit der stationären Phase wechselwirkt, jedoch innerhalb eines angemessenen Zeitraums eluiert, um die nachteiligen Auswirkungen der Bandenverbreiterung zu vermeiden.
Bei der Gradientenchromatographie sollten alle Peaks die gleiche Peakbreite aufweisen, da die sich ändernde Zusammensetzung der mobilen Phase das Band komprimiert. Wenn es bei der Gradientenchromatographie zu einer unerwarteten Änderung der Peakbreite kommt, ist dies häufig auf eine abgenutzte Säule zurückzuführen, die ausgetauscht werden muss.
Breite Peaks beeinträchtigen die Auflösung und Empfindlichkeit, insbesondere bei spät eluierenden Verbindungen in isokratischen Trennungen. Wie bereits erwähnt, kann eine zu hoch eingestellte Detektorempfindlichkeit zur Peakverbreiterung beitragen. Außerdem ist zu beachten, dass eine natürliche Alterung der Säule eine häufige Ursache darstellt.
Wenn es aufgrund von Dispersion im Injektionsventil zu einer Peakverbreiterung kommt, kann eine „Sandwich-Injektion“ mit kleinen Luftblasen dazu beitragen, diesen Effekt zu reduzieren. Eine weitere Ursache für Dispersion kann die Verwendung von Kapillaren mit einem zu großen Durchmesser oder die unbeabsichtigte Einführung von Totvolumen aufgrund von unsachgemäß festgezogenen Verbindungen sein.
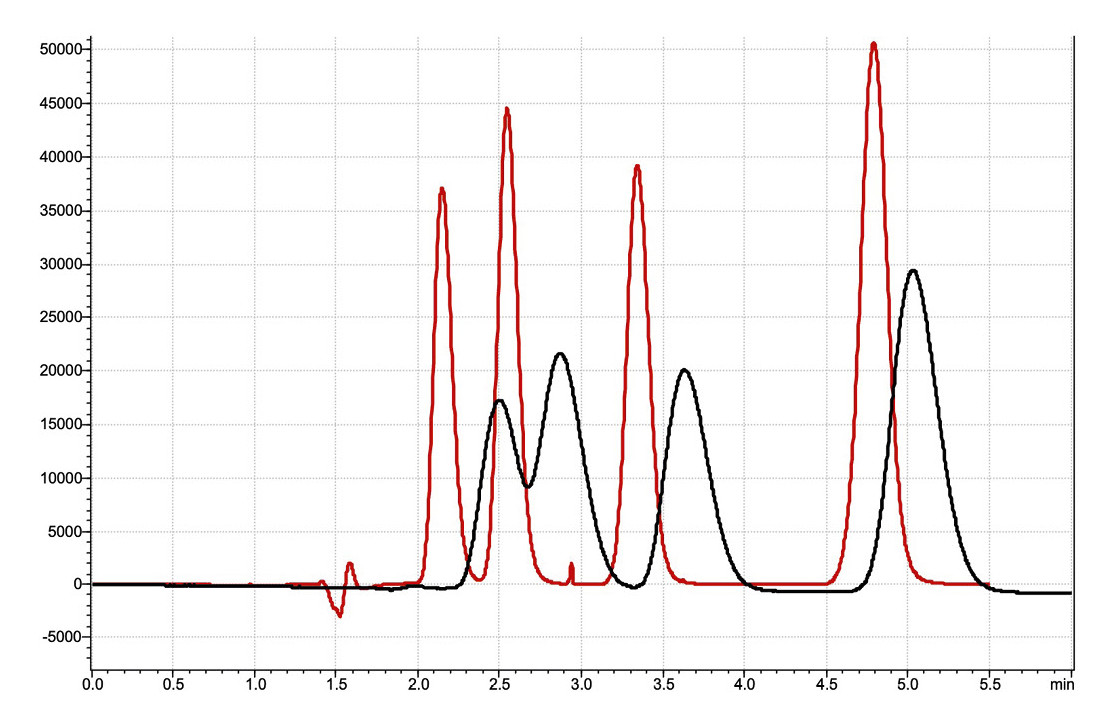
Was geschieht im rechten Chromatographen, wenn anstelle einer Kapillare mit einem Innendurchmesser von 0,125 mm eine breitere Kapillare mit einem Innendurchmesser von 0,5 mm verwendet wird? Die Baseline-Trennung geht verloren, und die Peaks sind verbreitert. Allerdings ist zu beachten, dass schmalere Kapillaren einen höheren Gegendruck im System erzeugen können. Daher müssen die Abmessungen der Kapillaren an das chromatographische System angepasst werden, damit die zulässigen Druckgrenzen nicht erreicht werden (Abbildung 7).
0,5 mm Kapillarinnendurchmesser (schwarz) statt 0,125 mm (rot) zwischen Säule und Detektor
Lösungen
- Die Verbindungen müssen überprüft und nachgezogen werden, um das Totvolumen zu minimieren.
- Es sollten Kapillaren verwendet werden, die für die Durchflussraten geeignet sind (unnötig große Innendurchmesser sind zu vermeiden).
- Die mobile Phase sollte vorgewärmt werden, und ein Säulenofen sollte verwendet werden, um eine gleichmäßige Temperatur aufrechtzuerhalten.
- Der Wechsel zu einer Gradientenelution kann die natürlichen Diffusionseffekte für späte Peaks reduzieren.
Zusammenfassung praktischer Richtlinien zur Vermeidung von Problemen mit der Peakform
- Die Zusammensetzung des Probenlösungsmittels sollte den anfänglichen Bedingungen der mobilen Phase entsprechen.
- Die verwendeten Injektionsvolumina sollten für die Kapazität der Säule geeignet sein.
- Die Aufnahmerate und Empfindlichkeitseinstellungdes Detektors sollten für die Methode optimiert werden.
- Die Säulen sollten sorgfältig behandelt und die Spezifikationen (pH-Wert, Temperatur) eingehalten werden.
- Das Systemvolumen sollte minimiert und alle Anschlüsse nach Hardwareänderungen überprüft werden.
- Die mobilen Phasen sollten vorgewärmt werden, um interne Temperaturgefälle zu vermeiden.
Gemeinsame Fehlerbehebung
Veränderungen der Peakform dienen häufig als Frühwarnzeichen für Probleme mit dem System, der Säule oder der Methode. Allmähliche Veränderungen deuten in der Regel auf eine Alterung der Säule hin, wohingegen plötzliche Verzerrungen häufig auf Hardware- oder Verfahrensfehler zurückzuführen sind. Durch die systematische Auseinandersetzung mit Lösungsmitteleinflüssen, Injektionstechniken, Detektorparametern und mechanischen Faktoren lassen sich die meisten Probleme mit der Peakform schnell diagnostizieren und beheben.
Eine wirkungsvolle Fehlerbehebung hängt von der Beobachtung, dem Vergleich mit Referenzdaten und der methodischen Eliminierung potenzieller Ursachen ab und führt letztendlich zur Wiederherstellung präziser, symmetrischer Peaks und zu einer zuverlässigen chromatographischen Leistung.
Auch der Austausch über Erfahrungen, Herausforderungen und Lösungen ist sinnvoll – genau darum geht es in dieser neuen Artikelserie. Shimadzu hat immer ein offenes Ohr für die Anliegen von Laboranwendern. Denn auch wenn die Arbeit im Labor bisweilen einsam erscheinen mag, ist sie doch Teil einer großen Gemeinschaft wissbegieriger Köpfe, die jeden Tag neue, wichtige Erkenntnisfelder erschließen. Die Fehlerbehebung ist nur ein Teil der Arbeit; daher ist es gut, sich daran zu erinnern, dass niemand diesen Weg allein gehen muss.
